Last week, I spent Thursday and Friday at the Strange Loop conference in St. Louis, Missouri. It turned out to be a pretty great conference, even if I missed out on a lot of interesting talks due to a school project I had to finish and a lot of time spent volunteering. The ones I did make it to were amazing, however. (You can access the slides for each of the talks at the Strange Loop presentations page).
Thursday
I started off with Hilary Mason‘s talk on machine learning, and even through I missed the first 10-15 minutes or so, I arrived in time to hear her (a) run through Bayes’ Theorem in about a minute without going too far over the audience’s heads (or so I thought), (b) give a high-level overview of her Twitter Commander (Github link) Python application (which uses machine learning techniques to filter tweets from the 640 people she follows on Twitter) (c) say “Cheat as much as you can” in regards to solving difficult problems (which is basically one of my own programming mantras) and (d) mention both of my machine learning textbooks (Machine Learning by Thomas Mitchell and Pattern Recognition and Machine Learning by Christopher Bishop) casually in the Q&A session after her talk. Not bad for only catching 45 minutes or so of her talk.
Then, I had to take a hiatus to finish up my project proposal for, ironically enough, my Machine Learning class. That took a while.
The next session I attended was by Kyle Simpson, better known as Getify in the webdev world. (I hadn’t heard of him, but it’s a big place and I’ve been out of the loop for a while.) He gave a talk promoting using Javascript for what he referred to as the “middle end” of webapp development–the part of the webapp where the browser code meets the server code. The basic idea is rooted in the DRY (Don’t Repeat Yourself) principle–rewriting code is bad, but when writing web applications, we typically use two different programming languages (one on the client, one on the server) and repeat ourselves all over the place. Getify’s premise is that since we can’t run PHP, ASP, or any other server-side language in the browser, we should change the server to run the browser’s language, Javascript. He actually built a proof of concept JS-based server from the V8 Javascript engine called BikehainJS (Github link). He built a basic, but functional URL shortener called shortie.me using BikechainJS and walked us through the basic code behind the app. Pretty cool stuff, and I like the idea, though I think he’s got a long way to go to get this off the ground.
The next talk I heard was on Clojure‘s solution to the Expression Problem by Chris Houser (or Chouser, as I knew him at Sentry Data Systems), co-author of the new The Joy of Clojure book. The Expression Problem is kind of complicated (I’m still not sure I understand all the nuances), but the basic idea is that when we try to make an existing data type do something it’s not meant to do (to “express” a new trait), there are major issues. Chouser’s example involved extending two custom classes (which we have complete control over) with methods to display themselves in a report, and then trying to back-port those display methods to built-in classes (such as Vectors in Java–see his talk for full details, because I’m glossing over a lot). Most languages provide some way to do this (wrapper classes, monkey patching), but Clojure has a couple of really cool methods called multimethods and protocols. Multimethods are a sort of function overloading while protocols feel like advanced monkey patching with some extra safeguards. While those methods are cool, I think I’ll stick with monkey patching since the languages I use most often don’t have those tricks and I haven’t run into any of monkey patching’s problems. (Yet.)
The last talk on Thursday was by Guy Steele, one of the original Lisp hackers and one of the original authors of the Jargon File, along with numerous other things. His talk was on parallel programming and how not to do it. After a long breakdown of a program he wrote on a punch card (which was fascinating and hugely relevant to the talk, but probably a little on the long side), he introduced a pretty simple idea: instead of manually mucking around with the details of parallel programming (analogous to what he had to do with his punch card), why don’t we just let the compiler figure it out? We already let the compiler (or really low level libraries) do most of the really annoying work like register management, anyway (unless we’re writing C or C++), and the compilers do a great job, much better than most people can do and tremendously faster. Why can’t we do that with parallel programming? We have to be a little smarter about how we design programs in the first place, and many (if not all) of the tricks we’ve been learning for the past 50 years or so no longer work, but the benefits are usually worth it in the long run, except for the smallest of toy programs. Once we’ve done the high-level work for the compiler, which it can’t do, we let it handle the nitty-gritty details of the memory and processor management. While Steele made his case, he also introduced his current work at Sun Labs: a programming language called Fortress where running code in parallel is almost trivial (so long as you design it right from the beginning, which is usually the problem). It does show a ton of potential and is really very cool–Fortress is definitely on my list of languages to check out.
Ok, that’s enough for now, as this post is getting quite long. I’ve covered all the talks I saw on Thursday, so now is a good time to take a break. I’ll finish up the talks from Friday later this week.


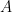 , eigenvector
, eigenvector  , and scalar
, and scalar  ,
, 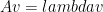
 is actually getting kind of fun, now that I’m figuring it out. I’ve even figured out how to draw simple diagrams and the like from within
is actually getting kind of fun, now that I’m figuring it out. I’ve even figured out how to draw simple diagrams and the like from within